- Motor system failure caused by progressive degeneration of UMNs (corticospinal and corticobulbar tracts) and LMNs (anterior horn cells)
- presents with upper or lower motor neurone signs, or a mixture of the two
- Progressive weakness of limbs, bulbar and respiratory muscles
- Does not involve sensory system
- incidence: approximately 2 per 100 000 per year except in endemic areas such as the Island of Guam
- prevalence: 5-6 per 100 000 of the population
- males affected more than females in ratio of 2:1
- familial link in 5-10% of cases
- age of onset – mean = 55 years; range = 16-77 years, but usually in fifth to seventh decades
- 3 subtypes
- Amyotrophic lateral sclerosis – ALS – most common – UMN and LMN signs, a late onset, rapidly progressive and ultimately fatal neurological disorder
- Progressive muscular atrophy – LMN features only
- Progressive bulbar – LMN in brainstem motor nuclei
- Primary lateral sclerosis – UMN signs only
- Clinical
- most patients visit their general practitioner first, typically with mild symptoms such as
- Cramps
- balance disturbance
- reduced dexterity
- Subtle cognitive changes including apathy
- time from symptom onset to diagnosis ranges from 10 – 16 months
- signs often go unrecognized, with patients referred to other specialists, or given misdiagnoses
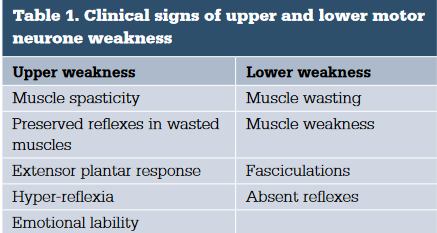
- Asymmetrical weakness in upper or lower limbs
- Weakness
- Mixed UMN / LMN signs
- brisk reflexes in a limb with muscle wasting
- Absence of sensory symptoms/pain
- Steadily progressive weakness
- Widespread fasiculations
- Preferential wasting lateral hand border
- Extra motor symptoms 50%
- cognitive and behavioural impairment
- language abnormalities
- Can have fronto-temporal dementiam – similar mechanisms
- Investigations
- If suspected needs referral to neurologist
- To exclude other causes MRI brain/spine
- Bloods including autoimmune screen, anti-ganglioside antibodies, ACH receptor antibodies, toxins, heavy metals, HIV, HIV , inflamamtory disease
- May get nerve conduction studies or EMG
- Counselling
- Incurable, usually death 3-5 years
- 90% have no family history, Often don’t find the gene
- Risk to first degree relatives 1-3% if not known to be familial
- Treatment
- Multi-disciplinary care
- Riluzole – protective for disease progression, survival benefit 3-6 months – monitor LFTs and neutropaenia monthly
- Early respiratory support – consider nocturnal NIV
- Maintain weight
Related